Benchmarks
Do you have a method you would like to use for studying photochemical reaction paths? Did you develop a new electronic structure method that you would like to test in different regions of a potential energy surface, including near-crossing regions? Are you interested in biological organic chromophores ? If so, you may be interested in testing your method on our Benchmark potential energy scans provided below.
In recent years, our group has produced a series of benchmark studies that apply a stringent test for electronic structure methods. These benchmarks are aimed at methods commonly used to study potential energy surfaces for photochemical reactions of organic chromophores, including surfaces near crossing regions. The purpose of this page is to make the geometries and tabulated results of these benchmarks publicly available for all who may be interested in testing their own method of choice (if we have not tested it already).
The Project was developed in our group within the activities of the IUPAC Subcommittee on Photochemistry and involved the contribution of the groups of:
Tadeusz Andruniów
Celestino Angeli
Andreas Dreuw
Nicolas Ferrè
Michael Filatov
Luis Manuel Frutos
Alexander Granovsky
Leonardo Guidoni
Miquel Huix-Rotllant
Anna Krylov
Roland Lindh
Donald Truhlar
The benchmark studies employing a reduced model of retinal protonated Schiff base (rPSB), the chromophore of visual pigments. This model, the Penta-2,4-dieniminium or 2,4-Pentadienylideneamonium cation (PSB3) features 3 double bonds but still preserves many of the features of the ground (S0) and first excited state (S1) potential energy surfaces of rPSB.
The benchmarks consist of a series of geometries tracing important pathways on the S0 and S1 surfaces of PSB3. These pathways follow reaction coordinates involved in the thermal and photochemical reactions of PSB3. More importantly, they pass through regions with changing electronic structure, and therefore provide a stringent test for electronic structure methods’ ability to describe these different pathways correctly. For more information, see the following references:
1.Gozem, S.; Huntress, M.; Schapiro, I.; Lindh, R.; Granovsky, A.A.; Angeli, C.; Olivucci, M. Dynamic Electron Correlation Effects on the Ground State Potential Energy Surface of a Retinal Chromophore Model. J. Chem. Theory Comput. 2012, 8, 4069-4080. http://dx.doi.org/10.1021/ct3003139.
2.Gozem, S.; Melaccio, F.; Lindh, R.; Krylov, A.I.; Granovsky, A.A.; Angeli, C.; Olivucci, M. Mapping the excited state potential energy surface of a retinal chromophore model with multireference and EOM-CC methods. J. Chem. Theory Comput. 2013, 9, 4495-4506. http://dx.doi.org/10.1021/ct400460h.
3.Gozem, S.; Krylov, A.I.; Olivucci, M. Conical Intersection and Potential Energy Surface Features of a Model Retinal Chromophore: Comparison of EOM-CC and Multireference Methods. J. Chem. Theory Comput. 2013, 9, 284-292. http://dx.doi.org/10.1021/ct300759z.
4.Xu, X.; Gozem, S.; Olivucci, M.; Truhlar, D. Combined Self-Consistent-Field and Spin-Flip Tamm Dancoff Density Functional Approach to Potential Energy Surfaces for Photochemistry. J. Phys. Chem. Lett. 2013, 4, 253-258. http://dx.doi.org/10.1021/jz301935x.
5.Huix-Rotllant, M.; Filatov, M.; Gozem, S.; Schapiro, I.; Olivucci, M.; Ferré, N. Assessment of density functional theory for describing the correlation effects on the ground and excited state potential energy surfaces of a retinal chromophore model. J. Chem. Theory Comput. 2013, 9, 3917-3932. http://dx.doi.org/10.1021/ct4003465.
6.Gozem, S. Melaccio, F., Valentini, A., Filatov, M., Huix-Rotllant, M., Ferre, N., Frutos, L. M., Angeli, C., Krylov, A.I., Granovsky, A. A., Lindh, R., Olivucci, M. Shape of Multireference, Equation-of-Motion Coupled-Cluster, and Density Functional Theory Potential Energy Surfaces at a Conical Intersection. J. Chem. Theory Comput., 2014, 10, 3074–3084. http://dx.doi.org/10.1021/ct500154k.
7.Schapiro, I.; Neese, F.. SORCI for photochemical and thermal reaction paths: A benchmark study Computational and Theoretical Chemistry 2014, 1040-1041, 84-98. doi:10.1016/j.comptc.2014.04.002
8.Zen A, Coccia E, Gozem S, Olivucci M, Guidoni L Quantum Monte Carlo Treatment of the Charge Transfer and Diradical Electronic Character in a Retinal Chromophore Minimal Model. J. Chem. Theory Comput., 2015, 11, 992-1005. http://dx.doi.org/10.1021/ct501122z.
9.Tuna, D.; Lefrancois D.; Wolański, Ł.; Gozem, S.; Schapiro I.; Andruniów, T.; Dreuw, A.; Olivucci M. Assessment of Approximate Coupled-Cluster and Algebraic-Diagrammatic-Construction Methods for Ground- and Excited-State Reaction Paths and the Conical-Intersection Seam of a Retinal-Chromophore Model J. Chem. Theory Comput., 2015, 11, 5758-5781. http://pubs.acs.org/doi/10.1021/acs.jctc.5b00022
10. Manathunga, M.; Yang, X.; Luk, H. L.; Gozem, S.; Frutos, L. M.; Valentini, A.; Ferre, N.; Olivucci, M. Probing the photodynamics of rhodopsins with reduced retinal chromophores Journal of Chemical Theory and Computation 2016, 12, 839-850. http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00945
These pathways have been used to test Multiconfigurational/ and Multireference methods (see refs. 1, 2 and 6), Equation-of-motion coupled cluster methods (refs. 2, 3 and 6), Density functional theory methods (refs. 4, 5 and 6), SORCI (ref. 7), Quantum Monte Carlo (ref. 8), and Approximate coupled cluster and Algebraic-Diagrammatic-Construction methods (ref. 9). The potential energy surfaces obtained with these methods are compared to those obtained at MRCISD+Q level of theory.
The structures used in these benchmark studies are provided via the links below. The paths themselves are minimum energy paths or interpolations/extrapolations of coordinates, which were either obtained at the CASSCF/6-31G* level of theory or CASPT2(IPEA=0.25)/6-31G* level of theory.
From ref. 1:
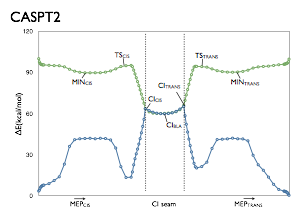
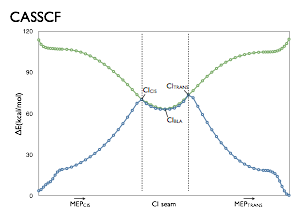
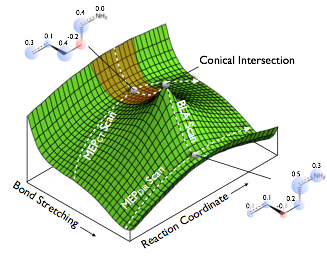
From ref. 2:
From ref. 2:

